hemophilia
Synonyms
Haemophilia, hereditary bleeding disorder, blood clotting factor deficiency,
Factor VIII deficiency, factor IX deficiency, hemophilia
definition
Hemophilia is an inherited disease of the blood clotting system:
The affected patients have impaired blood clotting, which manifests itself in the fact that bleeding occurs with the smallest trauma and the bleeding is difficult to stop.
Coagulation factors cannot be activated, so that the regular course of the coagulation cascade is not possible.
Hemophilia is a congenital disease that comes in two forms,
Hemophilia A and B, may be present.
Hemophilia A occurs more frequently (in 85% of cases) than form B (approx. 15%) and is the more serious of the two forms.
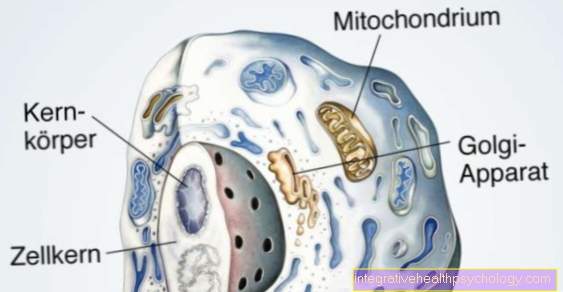
The activity of the factor VIII complex is limited in haemophilia A because the formation of factor VIII-C in the liver cells is reduced.
Factor VIII-C is one of the two subunits of the factor VIII complex: The von-Willebrand-Factor and factor VIII-C form this coagulation-promoting complex in the coagulation cascade. The von Willebrand factor protects factor VIII-C from being broken down too quickly.
The amount of functional factor VIII complex is consequently reduced in form A.
In the hemophilia B. the synthesis of coagulation factor IX takes place to a lesser extent, so that its effect in the coagulation process is absent and hemostasis is impaired.
Occurrence / epidemiology
The occurrence of hemophilia in the population is for haemophilia A. 1 in 5,000 and for form B. 1:15.000.
The fact that men in particular get sick is due to gender-related issues
X-linked form of inheritance of the coagulation defect (see cause / development).
Basics
Blood coagulation is a finely tuned system: Various components of the blood, the so-called coagulation factors, are activated one after the other in a cascade-like manner in order to enable hemostasis in the event of an injury.
The coagulation system consists of primary and secondary hemostasis, which is also called plasmatic coagulation.
If a vessel wall is injured, the affected vessel narrows in a first step (= primary coagulation). Then a prop (=Platelet thrombus) at the injured area, which is covered by platelets (= Platelets) is formed in order to achieve an initial seal of the vessel.
The formation of the thrombus activates the plasmastic coagulation:
The coagulation cascade starts and the interaction of the various coagulation factors creates a stable wound or vascular closure.
Cause and origin
The hemophilia Both forms of haemophilia are inherited as an X-linked recessive trait.
In the so-called X-linked mode of inheritance of hemophilia, the genetic information for a gene product is on the sex chromosome X, which is found in double numbers in women and in single numbers in men.
There are two genes for every genetic information, the so-called alleles. If there is recessive inheritance, both alleles must be affected by a mutation in order to cause the disease: Recessive means that the pathologically altered genetic information is derived from a healthy, i.e. does not change, information is suppressed and does not lead to disease. A recessive hereditary disease only develops if both alleles carry incorrect genetic information.
Is only a Gene expression changed, come it Not to hemophilia.
Info: inheritance
If only one gene expression is changed, there will be no disease.
Since men only have one X chromosome in the sex chromosome pair and therefore information for gene products on their X chromosome is only available in a simple form, they will fall ill if only one allele is defective.
Women, on the other hand, who have two X chromosomes as sex chromosomes, only become ill when both chromosomes have faulty genes for the gene product.
In men suffering from haemophilia, the allele that carries the hereditary information for coagulation factor VIII-C (haemophilia A) or coagulation factor IX (haemophilia B) is only available in a slightly modified form;
Women have to carry this genetic change on both alleles in order to be sick.
If women have a faulty and a non-changed allele, they are called carriers of an X-linked inherited disease such as haemophilia.
In haemophilia, the faulty genetic information has the effect of reducing the activity of the affected coagulation factor or of reducing the production of coagulation factor.
The X chromosome is the carrier of the genetic material for the coagulation factors for secondary hemostasis.
Approx. 70% of the mutations in the genetic material that cause haemophilia are passed on in families, 30% of the diseases arise from spontaneous changes (= spontaneous mutations) in the genetic information.
diagnosis
After asking about the patient's medical history and doing a thorough physical examination, there are additional steps in the diagnosis of hemophilia:
In 2/3 of the cases there are cases of haemophilia in the family, which is why a familial clustering of the disease should be asked if the patient presents to the doctor with symptoms suspicious of haemophilia.
The patients report bruises as a result of the smallest injuries.
One differentiates those Severity of the hemophilia into a mild, moderate or severe form of the disease.
Examination of a blood sample provides the following results typical for haemophilia:
The bleeding time is normal (= the primary coagulation is intact), but the function of the plasmatic coagulation is reduced, which is why the so-called PTT time is longer.
PTT stands for the partial thromboplastin time. It is measured in laboratory tests to check the function of factors I, II, V, VIII to XII as well as XIV and XV.
Since one factor in the coagulation cascade is missing in hemophilia, it cannot run in an optimal manner, which results in a prolonged coagulation time.
In order to be able to differentiate between haemophilia A and B, the patient's blood must be examined for factors VIII and IX; the absence of the two factors determines the type of hemophilia.
Differential diagnosis
Haemophilia must be differentiated from other disorders of the coagulation system, with the von Willebrand-Jürgens syndrome being mentioned in particular.
The von Willebrand factor (vWF) works together with the factor VIII-C im
Factor VIII complex and causes the inhibition of the premature factor VIII degradation, in addition, the factor promotes coagulation by mediating the adhesion of blood platelets to the injured vascular site. Thus, the vWF is an important part of both primary and secondary coagulation.
If the von Willebrand-Jürgens syndrome is present, then the formation of the
vWFs in the vessel wall cells only take place to a small extent or in an incorrect manner. The reduced rate of education or the inadequate function of the
von Willebrand factor is caused by mutations in the gene for factor formation. The inheritance of the syndrome or the causative gene mutation is inherited in an autosomal dominant manner: the genetic information for the vWF is on chromosome number 12, which is not a sex chromosome. In the dominant mode of inheritance, a diseased allele suppresses the effect of the second, healthy allele, so that the disease is caused by the mutation of a gene and causes clinical symptoms.
The patients typically suffer from skin and mucous membrane bleeding, less from spontaneous bleeding like the haemophilia patients.
The disease is often only noticed when there is prolonged bleeding after an operation.
The bleeding time of the patient after an injury is prolonged and the values of the plasmatic coagulation are pathologically changed (prolonged PTT).
The von Willebrand-Jürgens syndrome is caused by the administration of desmopressin or the replacement of factor VIII and vWF to stabilize the coagulation (for a more detailed explanation see under “Therapy of haemophilia”).
What are the symptoms of hemophilia?
The symptoms in the forms of hemophilia do not differ:
- The hemophilia usually only leads to clinical symptoms in men.
- Excessive bleeding occurs with hemophilia, which is not in relation to the previous, mostly banal accident (trauma), the bleeding time is normal, but bleeding typically occurs that does not occur in healthy people.
- A distinction can be made between three forms of hemophilia, which are defined by the residual activity of the affected coagulation factors:
1.severe hemophilia (hemophilia) with less than 1% residual activity or a proportion of the functional factor still present in which spontaneous bleeding occurs and joint bleeding occurs
2. Moderate hemophilia (blood disease) with factor activity ranging between
1 and 5% of the normal factor activity and the occurrence of bruises (= hematomas) after minor trauma
3. mild haemophilia (hemophilia) in which there is still 5-15% residual activity and in which patients report symptoms such as hematomas (bruises) after significant trauma and postoperative bleeding.
Read more on the topic: What are the causes of a cerebral hemorrhage
- Women who usually only have altered genetic information for the hemophilia usually show no clinical symptoms with over 50% residual activity.
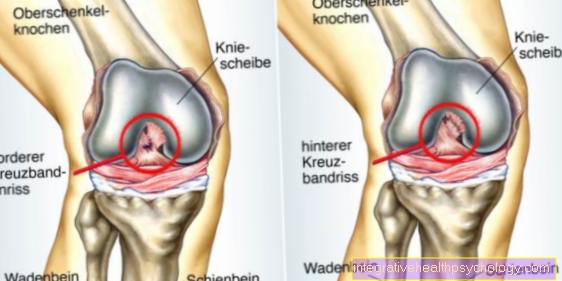
- Patients experience bleeding into large joints like that knee-, Hip- or Shoulder joint, whereby one speaks of hemarthrosis. The bleeding triggers an inflammatory reaction with repair processes in the joint, which can lead to a stiffening of the joint.
- It can also bleed into the Musculature and the soft tissues come. The bleeding leads to an increase in pressure in the muscles or the tissue, since there is now more volume. This can especially affect your arms and legs Compartment syndrome to lead:
The increased pressure causes the compression of blood vessels and nerves, so that the extremities are undersupplied and large areas of tissue can die. Compartment syndrome needs to be treated by the surgeon as soon as possible to prevent loss of the extremity. - Bleeding into the abdomen occurs, which is a life-threatening situation for the patient.
- Unusually long-lasting bleeding is possible after surgery.
In addition, there may be long periods with blood in the urine. Here one threatens Anemiabecause the patient is constantly losing blood, possibly unnoticed. - Are particularly dangerous Cerebral hemorrhage (= intracranial bleeding), which leads to death in 10% of people with haemophilia. You can find out more about this topic under our topic Cerebral hemorrhage.
How is therapy for hemophilia performed?

Bleeding should occur in the sick hemophilia must be avoided, which is why the patient does not Medication that inhibit blood clotting such as Acetylsalicylic acid (Aspirin®) and no intramuscular injections (= syringes into the Musculature) respectively.
If a trauma with bleeding occurs, careful local hemostasis is of great importance in order to prevent bleeding into neighboring tissue.
Drug therapy of the hemophilia consists in the replacement of coagulation factors that the body cannot produce in sufficient quantities.
Patients with a slight hemophilia receive the coagulation factor preparations as needed, i.e. if there has been trauma with bleeding or if major surgery is planned, which may result in profuse bleeding.
Prophylactic factor replacement should be used in patients with moderate to severe hemophilia, i.e. the missing factor should be given permanently, before bleeding occurs.
If there is residual activity of factor VIII of more than 15% or of factor IX of more than 20-25%, regular therapy is not necessary; these patients receive the missing coagulation factor in case of spontaneous bleeding or before planned surgery.
Long-term therapy as well as that before surgery can take the form of home self-treatment, in which the patient applies the missing coagulation factor himself.
There is a therapy alternative for patients with mild haemophilia:
The active substance Desmopressin (e.g. Minirin®) leads to a distribution of the
Factor VIII, which is stored in the vessel walls.
However, the drug can only be given for a few days at a time, since after stimulation of the factor release, the memory in the vessel walls is exhausted and has to be replicated again.
In acute therapy the hemophilia there are three options for intervention:
- The patient receives activated prothrombin, a substance that promotes blood clotting so that the blood is stopped as quickly as possible.
- Another therapeutic option is the administration of factor VII preparations. This factor is at the beginning of the coagulation cascade and initiates its regular course.
- The third therapeutic option is the application of animal factor VIII-C to enable the patient to stop bleeding.
Complications
The substitute gift (=substitution) of coagulation factors can lead to the formation of antibodies against precisely these factors, so that the substitution in a constant dose no longer has any therapeutic effects, which is why one speaks of the formation of inhibitors.
Depending on the determination of the concentration of the antibodies in the patient's blood, high doses of factor VIII can be administered, the aim of which is to regain tolerance to this factor and to switch off the formation of antibodies. This procedure should only be performed in specialized centers.
There is a low risk of contracting hepatitis or contracting the HIV virus (=HIV) as the factors are derived from human blood products.
Another risk of substitution therapy for hemophilia is the occurrence of allergic reactions.